


 Deutschland
Deutschland India
India Lietuva
Lietuva United Kingdom
United Kingdom Search
Search Search
Search


The societal benefits of medical innovation are clear—but the healthcare industry continues to face cost challenges as that innovation rapidly advances. Increasing complexity of care delivery, site of care needs, and adverse event monitoring requirements have been pushing more high-cost treatments into the inpatient hospital setting. With the Diagnosis Related Group (DRG) model providing a singular payment instead of a fee-for-service payment, institutions are facing increasing financial pressures.
Given the escalating complexity, cost, and resource needs of innovative medical advancements, all stakeholders face headwinds: the Centers for Medicare & Medicaid Services (CMS) in controlling costs; hospitals staying in business to provide care; and pharmaceutical manufacturers recouping costs to invest in future innovation. When administering complex new treatments for Medicare populations, hospitals often lose money. One example of this is with the introduction of CAR-T cell therapy treatments. As outlined in a 2021 Guidehouse-authored article published in Cell & Gene, CAR-Ts originally mapped to a DRG paying just $43,094—leading to hospitals losing significant margin when administering these products.
In response to manufacturers and advocacy groups lobbying for equitable payment, CMS established a new DRG 018, marking a critical milestone toward establishing adequate payment for novel, expensive treatment options. While CMS has indicated its openness to adjusting the DRG system—giving new complex therapies a much more favorable payment environment—it is still working to keep up with the rapid pace of innovation.
In April 2023, CMS released the Fiscal Year (FY) 2024 inpatient prospective payment system (IPPS) proposed rule, which included a record number of new technology add-on payment (NTAP) applications. CMS noted that applications have increased from a range of two to 10 per year in the first 20 years since the NTAP program was introduced, to 54 applications for FY 2024—a 200% increase from FY 2020. This demonstrates the continued need for DRG payment rates to keep up with rapidly advancing and costly treatments. Notably, in the IPPS final rule released August 2023, about 41% of those applications were approved.
Total NTAP Applications by Technology Type

50% of all FY 2020-2024 applications* were from pharma (Drugs and Biologics)
*Does not include 37 applications that were withdrawn prior to publication in the IPPS proposed rule. Also excluded from the data were New COVID-19 Treatments Add-on Payment applications, effective from November 2, 2020, to May 11, 2023.
NTAP Outcomes for Drugs and Biologics

*Includes one drug and device combo product with premarket approval.
While the majority (87%) of pharma-submitted drug and biologic NTAP applications were approved in the last five years, most applications were critically evaluated against CMS’s criteria of Newness, Cost, and Substantial Clinical Improvement (SCI).
Drug and Biologic Denial Reasons

Among the 13% of denied drug and biologic applications, most were unable to successfully make their case for their product being a substantial clinical improvement over other available therapies. CMS often publishes in the annual IPPS proposed rules many concerns over the limitations of published literature submitted in support of meeting the SCI criterion. In many cases of denied applications, applicants have been unable to provide both sufficient evidence and reasonable explanations for their limitations through the proposed rule comment period. These are summarized in the IPPS final rules and responded to by CMS.
NTAP Outcomes for Devices

*Includes a Drug and Device combo product with premarket approval and two software-based technologies.
Historically, NTAP applications for devices have had the most denials, averaging 36% over the past five years. But the advent of the alternative pathway for breakthrough devices in 2021 has dramatically increased the number of approvals, representing 86% of all device applications. This is made possible by having both the Newness and SCI criteria waived when the device has received the U.S. Food and Drug Administration (FDA) Breakthrough Device designation for the same indication when receiving FDA marketing authorization.
Device Denial Reasons

For device applications reviewed under the traditional NTAP pathway, a majority of denials were due to not meeting SCI alone (69%) or SCI and Newness combined (13%).
Total Annual NTAP Applications
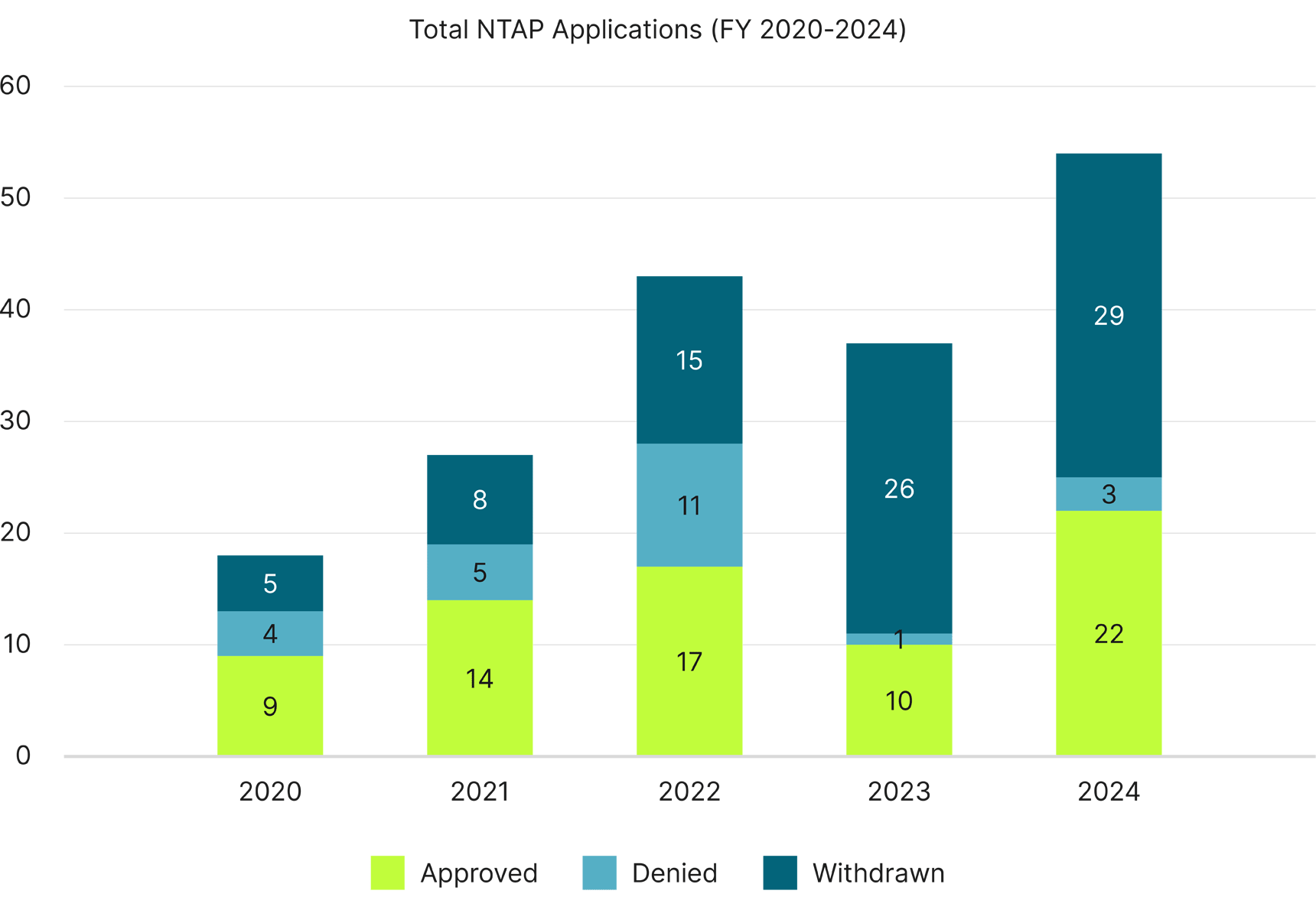
With the exception of FY 2023, NTAP approvals overall have increased year-to-year, with FY 2024 having the most NTAP applications in history and more than twice as many approvals as in FY 2020.
In an effort to address increasing application complexity and lack of critical information available to properly evaluate technologies for NTAP within the timeframe of the rule cycle (proposed and final rules), CMS has implemented changes to NTAP criteria beginning with the FY 2025 NTAP application cycle.
Manufacturers will be required to include proof of FDA filing with their NTAP submission, which typically is due by mid-October. The FDA filing must also have a “pending” or “in review” status, not a “delayed” or “hold” status. And the technology must receive FDA market authorization by May 1 of the year following NTAP application submission rather than the previous July 1 deadline.
The requirement to submit proof of FDA filing with NTAP applications will likely work to achieve the agency’s goal of having applications include information more closely aligned with the FDA label. With the expectation of earlier filing compared to the NTAP application deadline, CMS will have more time to properly evaluate applications for the proposed rule that’s published in April but drafted months earlier for review by the U.S. Office of Management and Budget.
The new NTAP requirements could lower the annual volume of NTAP applications due to the 7.5-month window from NTAP submission in mid-October to the new marketing authorization deadline of May 1. An NDA filing with the FDA typically takes about six to 10 months from the initial FDA application request to receive an approval decision.1,2 Unless manufacturers are filing well in advance of the NTAP application deadline, the number of withdrawals due to not meeting the May 1 deadline may be similar to recent years.
Future IPPS rule cycles may see a proposal to change the evaluation criteria of approved NTAPs for another year of eligibility—NTAP extending if the three-year anniversary of FDA approval or market availability falls within the second half of the next fiscal year—to conform with the maximum period for NTAP designation of two to three fiscal years from FDA approval or market availability; a policy change has potential to mitigate the impact on the application’s FDA approval deadline changing to two months earlier (May 1 vs. July 1). This would address comments summarized in the FY 2024 final rule about the three-year NTAP eligibility restriction.
While NTAP and outlier payments may provide early benefits to hospitals seeking to provide access to innovative technologies, these are not a long-term solution. Ensuring beneficiary access to innovative care while controlling overall costs is a complex issue and will require continued collaboration between CMS, hospitals, and life sciences innovators.
To successfully launch new technologies and therapies in this restricted payment environment, manufacturers need to develop robust reimbursement strategies that incorporate three key tactics.
1. Ensure early understanding of site of care exposure.
Pharmaceutical and medical device manufacturers need to develop early forecasts on anticipated site of care mix for their forthcoming therapies, because this has direct implications for reimbursement. Under today’s healthcare payment system, procedures are paid for differently depending on where the service occurs. While there are risks and opportunities involved for payment across all sites of care, the inpatient reimbursement system requires careful assessment and preparation in order to ensure that providers are paid adequately for cutting-edge treatments. By ensuring a strong understanding of inpatient site of care exposure early on, manufacturers can develop strong reimbursement strategies that successfully navigate the complexities of inpatient payment across books of business, such as determining appropriate codes, understanding procedure costs, and evaluating NTAP potential.
2. Design an evidence package that meets CMS payment policy milestones.
FDA approval does not guarantee coverage and payment. Because CMS and commercial payers alike often set higher evidence standards for securing access and payment, manufacturers must carefully consider their market access strategy before finalizing clinical trial designs. Designing a comprehensive evidence package that meets CMS requirements for key coverage and reimbursement policy milestones, such as the NTAP criteria, is critical to optimizing reimbursement potential.
3. Educate hospital customers on the costs associated with new technologies to ensure that future reimbursement accounts for the value of innovation.
A strong educational partnership at the account level is crucial to ensure that hospitals can onboard, treat, and adequately charge—commensurate to cost—for novel therapies. In the early years of a new procedure, CMS collects hospital costs through charge data in order to inform formal reimbursement rates. Providing early adopters with the appropriate resources to efficiently code, calculate costs, and subsequently charge to CMS helps more accurately account for the value of innovation—reducing the prospect of future payment erosion arising from miscalculation of appropriate payment rates.
Product development milestones are just one step along the broader journey of securing adequate reimbursement—and reimbursement and pricing considerations must be uniquely adapted to product- and company-specific needs. As DRG payment continues to exert pressure on inpatient hospitals, it will be critical for manufacturers to set up sustainable pricing and reimbursement policy strategies that facilitate access to innovative therapies for the patients who need them most.
Chance Scott, Partner
Guidehouse is a global advisory, technology, and managed services firm delivering value to commercial businesses and federal, state, and local governments. Serving industries focused on communities, energy, infrastructure, healthcare, financial services, defense, and national security, Guidehouse positions clients for AI-led innovation, efficiency, and resilience.